На этом шаге мы рассмотрим особенности применения PCA.
Одним из наиболее распространенных применений PCA является визуализация высокоразмерных наборов данных. Как мы видели ранее, довольно сложно построить диаграммы рассеяния для данных, которые включают больше двух признаков. Для набора данных Iris мы смогли построить матрицу диаграмм рассеяния, которая дала нам частичное представление о данных, показав все возможные комбинации двух признаков. Но если мы захотим взглянуть на набор данных Breast Cancer, использование матрицы диаграмм рассеяния будет затруднительным. Этот набор данных содержит 30 признаков, которые привели бы к 30 * 14 = 420 диаграммам рассеяния! Мы никогда не сможем детально просмотреть все эти графики, не говоря уже об их интерпретации.
Впрочем, можно воспользовать более простой визуализацией, вычислив гистограммы распределения значений признаков для двух классов, доброкачественных и злокачественных опухолей (рисунок 1):
[In 14]: fig, axes = plt.subplots(15, 2, figsize=(10, 20)) malignant = cancer.data[cancer.target == 0] benign = cancer.data[cancer.target == 1] ax = axes.ravel() for i in range(30): _, bins = np.histogram(cancer.data[:, i], bins=50) ax[i].hist(malignant[:, i], bins=bins, color=mglearn.cm3(0), alpha=.5) ax[i].hist(benign[:, i], bins=bins, color=mglearn.cm3(2), alpha=.5) ax[i].set_title(cancer.feature_names[i]) ax[i].set_yticks(()) ax[0].set_xlabel("Значение признака") ax[0].set_ylabel("Частота") ax[0].legend(["доброкачественная", "злокачественная"], loc="best") fig.tight_layout()
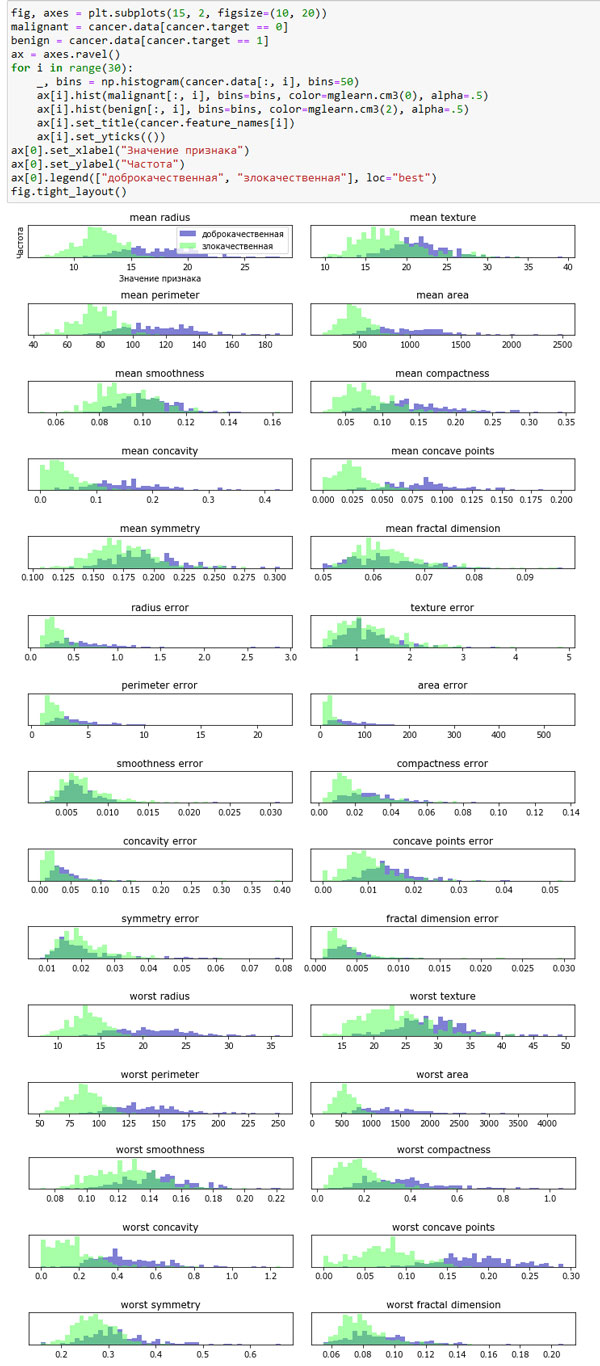
Рис.1. Преобразование данных с помощью РСА
В данном случае мы строим для каждого признака гистограмму, подсчитывая частоту встречаемости точек данных в пределах границ интервалов (этот интервал еще называют бином). Каждый график содержит две наложенные друг на друга гистограммы, первая - для всех точек, относящихся к классу "доброкачественная опухоль" (синий цвет), а вторая - для всех точек, относящихся к классу "злокачественная опухоль" (зеленый цвет). Это дает нам некоторое представление о распределении каждого признака по двум классам и позволяет нам строить предположения о том, какие признаки лучше всего дискриминируют злокачественные и доброкачественные опухоли. Например, признак "smoothness error", похоже, довольно малоинформативен, потому что две гистограммы, построенные для данного признака, большей частью накладываются друг на друга, в то время признак "worst concave points" кажется весьма информативным, поскольку гистрограммы, построенные для этого признака, практически не накрывают друг друга.
Однако этот график не дает нам никакой информации о взаимодействии между переменными и взаимосвязях между признаками и классами зависимой переменной. Используя PCA, мы можем учесть главные взаимодействия и получить несколько более полную картину. Мы можем найти первые две главные компоненты и визуализировать данные в этом новом двумерном пространстве с помощью одной диаграммы рассеяния.
Перед тем, как применить PCA, мы отмасштабируем наши данные таким образом, чтобы каждый признак имел единичную дисперсию, воспользовавшись StandardScaler:
[In 15]: from sklearn.datasets import load_breast_cancer cancer = load_breast_cancer() scaler = StandardScaler() scaler.fit(cancer.data) X_scaled = scaler.transform(cancer.data)
Обучение PCA и его применение так же просто, как применение преобразований, выполняющихся в ходе предварительной обработки. Мы создаем экземпляр объекта PCA, находим главные компоненты, вызвав метод fit(), а затем применяем вращение и снижение размерности, вызвав метод transform(). По умолчанию PCA лишь поворачивает (и смещает) данные, но сохраняет все главные компоненты. Чтобы уменьшить размерность данных, нам нужно указать, сколько компонент мы хотим сохранить при создании объекта PCA:
[In 16]: from sklearn.decomposition import PCA # оставляем первые две главные компоненты pca = PCA(n_components=2) # подгоняем модель PCA на наборе данных breast cancer pca.fit(X_scaled) # преобразуем данные к первым двум главным компонентам X_pca = pca.transform(X_scaled) print("Форма исходного массива: {}".format(str(X_scaled.shape))) print("Форма массива после сокращения размерности: {}".format(str(X_pca.shape))) Форма исходного массива: (569, 30) Форма массива после сокращения размерности: (569, 2)
Теперь мы можем построить график первых двух главных компонент (рисунок 2):
[In 17]: # строим график первых двух главных компонент, классы выделены цветом plt.figure(figsize=(8, 8)) mglearn.discrete_scatter(X_pca[:, 0], X_pca[:, 1], cancer.target) plt.legend(cancer.target_names, loc="best") plt.gca().set_aspect("equal") plt.xlabel("Первая главная компонента") plt.ylabel("Вторая главная компонента") Text(0, 0.5, 'Вторая главная компонента')
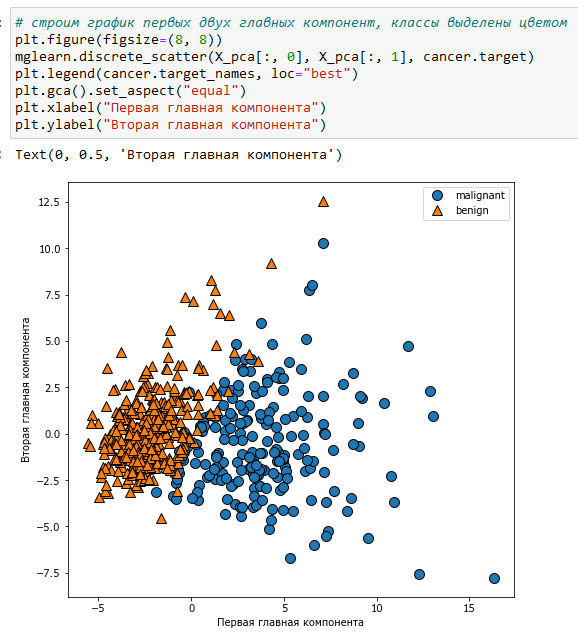
Рис.2. Двумерная диаграмма рассеяния для набора данных Breast Cancer с использованием первых двух главных компонент
Важно отметить, что РСА является методом машинного обучения без учителя и не использует какой-либо информации о классах при поиске поворота. Он просто анализирует корреляционные связи в данных. Для точечного графика, показанного здесь, мы построили график, где по оси x отложена первая главная компонента, по оси у - вторая главная компонента, а затем воспользовались информацией о классах, чтобы выделить точки разным цветом. Вы можете увидеть, что в рассматриваемом двумерном пространстве эти два класса разделены достаточно хорошо. Это наводит на мысль, что даже линейный классификатор (который проведет прямую линию в этом пространстве) сможет достаточно хорошо разделить два класса. Кроме того, мы можем увидеть, что случаи злокачественных опухолей (синие точки) более распространены, чем случаи доброкачественных опухолей (зеленые точки) - что отчасти было видно на гистограммах рисунке 1.
Недостаток PCA заключается в том, что эти две оси графика часто бывает сложно интерпретировать. Главные компоненты соответствуют направлениям данных, поэтому они представляют собой комбинации исходных признаков. Однако, как мы скоро увидим, эти комбинации обычно очень сложны. Сами главные компоненты могут быть сохранены в атрибуте components_ объекта PCA в ходе подгонки:
[In 18]: print("форма главных компонент: {}".format(pca.components_.shape)) форма главных компонент: (2, 30)
Каждая строка в атрибуте components_ соответствует одной главной компоненте и они отсортированы по важности (первой приводится первая главная компонента и т.д.). Столбцы соответствуют атрибуту исходных признаков для объекта PCA в этом примере, "mean radius", "mean texture" и т.д. Давайте посмотрим на содержимое атрибута components_:
[In 19]: print("компоненты PCA:\n{}".format(pca.components_)) компоненты PCA: [[ 0.21890244 0.10372458 0.22753729 0.22099499 0.14258969 0.23928535 0.25840048 0.26085376 0.13816696 0.06436335 0.20597878 0.01742803 0.21132592 0.20286964 0.01453145 0.17039345 0.15358979 0.1834174 0.04249842 0.10256832 0.22799663 0.10446933 0.23663968 0.22487053 0.12795256 0.21009588 0.22876753 0.25088597 0.12290456 0.13178394] [-0.23385713 -0.05970609 -0.21518136 -0.23107671 0.18611302 0.15189161 0.06016536 -0.0347675 0.19034877 0.36657547 -0.10555215 0.08997968 -0.08945723 -0.15229263 0.20443045 0.2327159 0.19720728 0.13032156 0.183848 0.28009203 -0.21986638 -0.0454673 -0.19987843 -0.21935186 0.17230435 0.14359317 0.09796411 -0.00825724 0.14188335 0.27533947]]
Кроме того, с помощью тепловой карты (рисунок 3) мы можем визуализировать коэффициенты, чтобы упростить их интерпретацию:
plt.matshow(pca.components_, cmap='viridis') plt.yticks([0, 1], ["Первая компонента", "Вторая компонента"]) plt.colorbar() plt.xticks(range(len(cancer.feature_names)), cancer.feature_names, rotation=60, ha='left') plt.xlabel("Характеристика") plt.ylabel("Главные компоненты") Text(0, 0.5, 'Главные компоненты')
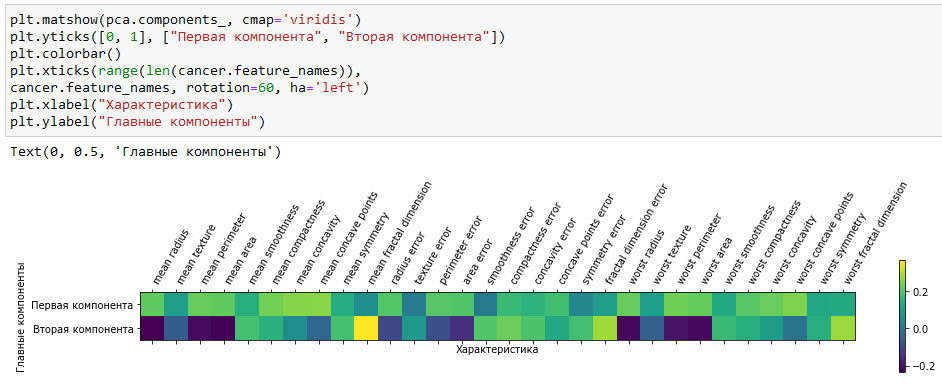
Рис.3. Тепловая карта первых двух главных компонент для набора данных рака Breast Cancer (изображение кликабельно)
Вы можете увидеть, что в первой компоненте коэффициенты всех признаков имеют одинаковый знак (они положительные, но, как мы уже говорили ранее, не имеет значения, какое направление указывает стрелка). Это означает, что существует общая корреляция между всеми признаками. Высоким значениям одного признака будут соответствовать высокие значения остальных признаков. Во второй компоненте коэффициенты признаков имеют разные знаки. Обе компоненты включают все 30 признаков. Смешивание всех признаков - это как раз то, что усложняет интерпретацию осей на рисунке 3.
На следующем шаге мы рассмотрим метод "собственных лиц" (eigenfaces) для выделения характеристик.
